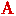自从1925年H.S Taylor提出催化活性中心的概念以来,人们就试图通过各种方法与理论理解催化活性的起源,以期能够快速而准确地预测活性中心的位置,并达到设计高活性催化材料的目的。这其中,基于中间物吸附/脱附以及d带中心等理论的密度泛函理论计算取得了巨大的成功。但是这种方法算量巨大,对算力和人力都提出了极高的要求,此外也很难对材料的所有晶面做出全面的分析,极大限制了其对新材料的预测功能。那么,是否有一种理论,可以绕过复杂的第一性原理计算,实现催化活性中心的快速判断呢? 近年来,拓扑能带理论的发展为高通量预测材料的电子结构提供了新的机遇。以拓扑量子化学理论(topological quantum chemistry)为代表(Nature, 2019, 566, 480),由于晶体能带可由空间群的不可约表示来标记,因此可以从晶体对称性出发,对材料的电子结构和性质做出预测。近期,中国科学院宁波材料技术与工程研究所磁性相变材料团队李国伟研究员在系统研究拓扑表面电子态表面化学效应的基础上(npj Computational Materials, 2021, 7, 1; Applied Physics Letters, 2020, 116, 070501;Science Advances, 2019, 5, eaaw9867;Angewandte Chemie, 2019, 131, 13241),与马普固体物理化学研究所Claudia Felser教授及普林斯顿大学Andrei Bernevig教授合作,通过扫描无机晶体数据库中数万个拓扑平庸材料,确定了一种来源于材料体相的本征表面电子态—Obstructed surface states(OSSs),并可以用来快速准确判定无机晶体材料催化活性中心的可能位置。 研究团队从无机晶体数据库中的3万余种非拓扑材料出发(arXiv:2111.02433),计算了这些材料的实空间不变量(Real-space invariants),发现存在着这样一类材料,即obstructed Wannier电荷中心(OWCCs)与晶体的原子位置不重合时,会出现一种内禀的金属表面电子态(OSSs)。分析发现,这些金属电子态完美满足了催化材料活性位点的基本要求,即:当解离出该晶面时,会有表面态产生;具有良好的导电性;电子态位于费米面附近,易参与电子转移与成键。 为了证实这一猜想,他们以多种经典催化制氢材料为例,合成了肉眼可分辨不同晶体面的大尺寸单晶,证实了催化活性位点与OSSs所在晶面高度重合。以层状范德华材料MoX2(X=S, Se, Te)为例,理论表明,此类材料的OWCCs位于晶体Wyckoff位置2b处,这也预示着表面电子态不在晶体(001)位置,而是位于所有的垂直于(001)晶面的侧面内。这与众多的实验结果相符,即催化活性来源于非(001)晶面的侧晶面。通过导电探针原子力显微镜,他们发现导电性在晶体的边缘位置明显高于无OSSs存在的惰性面。更重要的是,可以通过考虑晶体对称性及OWCCs所在位置,判定OSSs存在的几率并用于预测潜在的高活性催化材料。 借助于这一机理,他们发现了3000余种类似的非磁性材料与30种磁性材料。考虑晶体化学稳定性及合成上的可能性及成本,从中预测了超过400种潜在的高活性催化材料(详见论文Appendix A),并详细给出了OSSs所在的Miller指数面。这证实了OSSs可以作为寻找催化活性中心的“描述符”,为快速判定活性位点位置、进而针对不同的催化场景设计高效催化材料提供了指导。 这一成果以“Obstructed Surface States as the Descriptor for Predicting Catalytic Active Sites in Inorganic Crystalline Materials”为题发表在权威期刊Advanced Materials(论文信息:doi.org/10.1002/adma.202201328)上,并被编辑遴选为当期卷首插画(Frontispiece)论文。第一作者及通讯作者是中科院宁波材料所李国伟研究员,马普固体微结构所许远锋博士为共同一作。此外,通讯作者还包括普林斯顿大学B. Andrei Bernevig教授以及马普固体物理化学研究所Claudia Felser教授。中科院宁波材料所刘剑研究员、张钰笛为该论文共同作者。该研究得到了中科院宁波材料所“团队人才”项目的支持,作者同时感谢其获得的第一个竞争性经费“所长基金科研项目-青年项目”的支持。 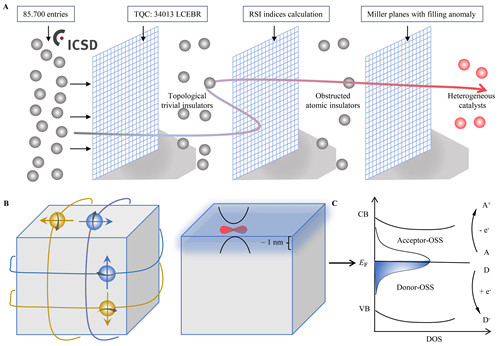 图1 基于拓扑量子化学理论的催化材料搜寻与设计思路  图2 通过本征表面态快速确定催化活性中心所在位置 (磁材实验室 李国伟)
|